











Research
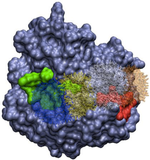
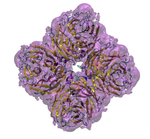
Our lab uses theoretical and computational techniques to investigate the interactions and dynamics of biological macromolecules. We hope to help elucidate the mechanisms by which these marcromolecules utilize intramolecular motion and intermolecular forces to achieve their biological functions. Currently, our main focus has been on exploring the pathways and kinetics of ligands binding to the influenza protein neuraminidase (Figure 1).
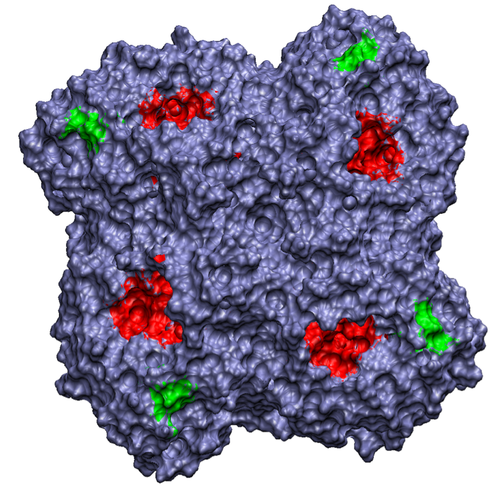
Figure 1. Influenza neuraminidase tetramer; active sites
are shown in red, secondary sites are shown in green.
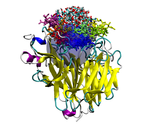
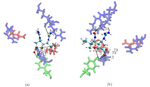
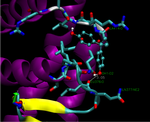
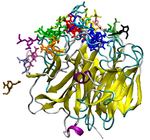
We are currently developing a computational methodology to accurately measure the association rate constants of small ligands to both the active (red in Figure 1) and secondary (green in Figure 1) binding sites. This methodology models the diffusional character of the association pathway using Brownian dynamics (BD) and the local, van der Waal contact search with classical molecular dynamics (MD) simulations. As shown in Figure 2, we are currently attempting to implement this multi-scale method by switching from the approximate BD to the more detailed MD at a proximal elliptical surface. The qualitative application of this methodology has allowed us to computationally predict the pathways of various ligands binding to neuraminidases from a variety of sources.
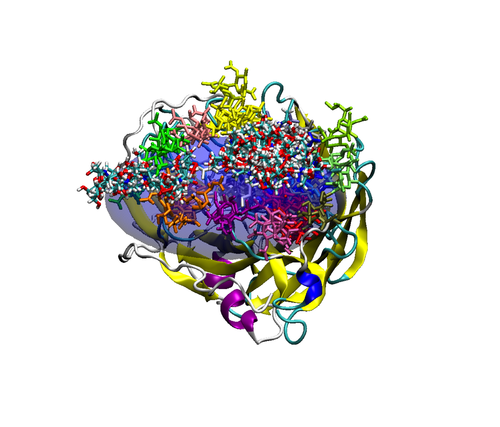
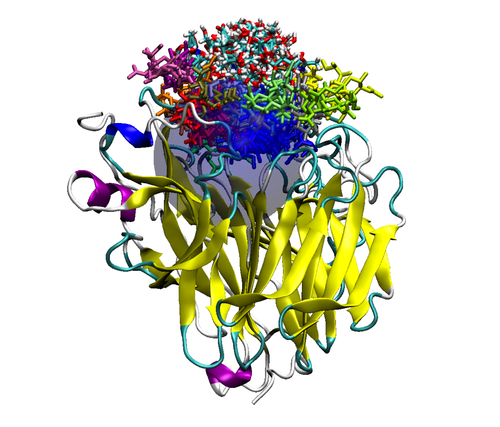
Figure 2. Top (left panel) and side (right panel) view of a neuraminidase monomer. The elliptical surface to switch between BD and MD is shown as partially
transparent. Ligands in atom-type coloring are BD arrivals to the transition surface; ligands in single colors are MD bound to the surface of the protein.